The IT/Clinical Engineering Governance Gap
July / August 2012
![]()
The IT/Clinical Engineering Governance Gap
Is your organization ready to support safety-critical systems?
With certain patient safety issues, the “system” is responsible for the root cause of a problem rather than the actions of those participating in the situation resulting in an adverse event. Clinical activities to which “the system” may contribute risk include patient handoffs between clinicians, medication administration, diagnosis of disease, and many others.
A system-based patient safety risk that has been emerging in the healthcare industry for some time—mostly unrecognized and inadequately addressed—is a gap in the governance of automated technologies in hospitals. This emerging risk is a consequence of progress in automating healthcare workflows and processes. Automation has moved beyond patient accounting, order entry, and results reporting to embrace remote surveillance, alarm notification, smart pumps, and clinical decision support systems. These systems meet the legal definition of a medical device, are regulated by the Food and Drug Administration (FDA), and can represent considerable risk to patient safety. The systemic patient safety risk arises from how these medical device systems are managed and supported. The relevant systems can be divided between medical device systems and medical device software.
Medical device systems are conventional medical devices connected via networks and server applications to extend the capabilities of standalone medical devices. Examples include patient monitoring systems that provide remove surveillance, alarm notification, retrospective event review, and data analysis. Smart pumps utilize networking and server-based applications to provide drug error reduction systems (DERS) to minimize errors in infused therapy administration. Beyond these widely adopted medical device systems are new systems targeting vital-signs monitors, ventilators, diagnostic point-of-care testing devices, and other point-of-care medical devices.
Software that makes or proposes a diagnosis or plans or controls a therapy often qualifies as an FDA-regulated medical device. Examples of medical device software include numerous diagnostic systems that acquire and analyze medical device data such as picture archiving and communications systems (PACS) and clinical decision support systems for applications such as tight glycemic control or the diagnosis and treatment of sepsis.
Historically, medical device systems were physically separate systems that were installed, serviced, and supported by the medical device manufacturer. Such standalone systems are “islands of information” and make it difficult to share information with other information systems. Medical device systems implemented as physically separate systems can also duplicate existing IT infrastructure. Hospitals’ substantial investments in IT infrastructure make it hard to justify duplicate infrastructure for medical device systems. Consequently, the manufacturer-supported standalone paradigm is starting to break down, and medical device systems are being deployed on hospital enterprise IT infrastructure in greater numbers. Medical device software also exists on enterprise IT infrastructure.
Medical device systems and medical device software are designed, tested, and cleared by FDA with detailed specifications and operating environments. The IT infrastructure used by the medical device system is as much a part of the regulated medical device as the medical device itself.
A change in a medical device systems’ operating environment—medical device firmware releases; access points; firmware releases in controllers, switches, and routers; operating system patches or upgrades; or virtualized environment changes—can result in medical device system failures. This is not an uncommon occurrence with diagnostic systems. Fortunately the risks associated with diagnostic system failures most often render the diagnostic system inoperable, causing delays rather than posing an active risk to patient safety. Similar failures of patient care devices used at the point of care to monitor patients and deliver therapy can and do represent active risks to patient safety.
Upon installation, medical device manufacturers insist that their medical device systems meet the operating environment specifications for which they were designed (and cleared by FDA for sale). This often requires hospitals to make adjustments to their IT infrastructure to accommodate these medical device systems (Figure 1, on page 24). Once a medical device system or medical device software is installed and “certified” by the medical device manufacturer as meeting specifications, the ongoing operation and maintenance of the system is turned over to the hospital. This is true even if the hospital has secured a maintenance-and-support agreement with the manufacturer because it is the hospital that controls the day-to-day management of their IT infrastructure.
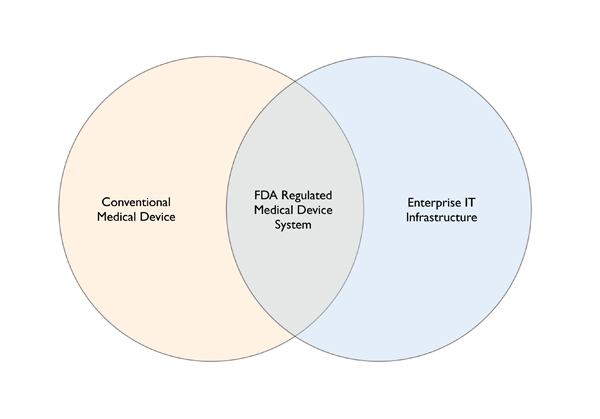
Figure 1. Convergence of Medical Device Systems and IT
The IT components of medical device systems have created an overlap between clinical engineering and IT. The converged area represents IT components and infrastructure that effectively becomes part of FDA-regulated medical device systems. Medical devices, including those portions that are part of IT infrastructure, represent safety-critical systems.
Operating these systems using different specifications or operating environments typically voids the medical device manufacturer’s warranty. Making any changes that have not been tested and validated by the medical device manufacturers results in the customer modifying a regulated medical device, creating patient safety risk and exposing the hospital to additional liability.
Clinical or biomedical engineering and IT are the two hospital departments principally responsible for managing medical device systems and medical device software. Typically, both departments are organized as if their two spheres of responsibility do not intersect. It is this intersection that represents an emerging patient safety risk. The current trend to have clinical engineering report to IT does not by itself mitigate this emerging risk. Should the concerns of clinical engineering be sufficiently discounted by IT management, patient safety risk could increase significantly.
Systemic risks arise from the nature of the relationship between clinical engineering and IT. Clinical engineers think of themselves as being responsible for medical devices, while IT is focused on enterprise information technology. Neither group has yet to really come to grips with the fact that increasingly, medical devices are information appliances that operate within the enterprise IT infrastructure. In the vast majority of hospitals, clinical engineering and IT work together effectively, yet in many of these cases there remain significant gaps in how medical device systems are managed and supported.
The best way to manage an asset is to enter it into an inventory so as to actively manage the support, upgrade, or repair of the asset over time. Medical device systems are most often captured in two separate inventories. The clinical engineering inventory is focused on the conventional medical device “box” or embedded system device, while the IT infrastructure, especially network components, are included in the IT inventory. Medical device system applications, servers, and client devices are most often included in IT’s inventory, although these components are sometimes found in clinical engineering inventories. Medical device software that does not include conventional medical devices can be found in either clinical engineering or IT inventories, or a combination of the two. The patient safety risk arises from the fact that the inventory of a medical device system is divided between two inventory systems, and there is rarely an audit to ensure that all inventory items are included in at least one inventory. Further, there is typically no way to manage a system’s components when they are split between two different inventory systems. Managing large, widely deployed medical device systems like patient monitoring or smart pump systems is further complicated when individual components are managed, but those components cannot be identified as parts of a particular medical device system.
Besides separate inventories, clinical engineering and IT have their own policies and procedures. Because an accreditation body occasionally audits them, clinical engineering typically has a more complete and formalized set of policies and procedures than IT does. Unfortunately, accreditation body requirements also lag with regard to medical device systems and are focused on discreet standalone medical devices; they do not directly address systems issues. Very few IT departments in hospitals today have fully implemented recognized policy and procedure frameworks such as ITIL1, IEC/ISO 200002 or COBIT3.
The separate policies and procedures between clinical engineering and IT frequently result in policy gaps, some of which are a consequence of being two separate departments and some which can be attributed to an inadequate awareness of medical device systems among both clinical engineering and IT. Key policy gaps often include risk management, configuration management, change control, and proper test environments.
Risk Management
When medical devices were limited to discreet standalone medical devices, hospitals rightly depended on manufacturers to complete adequate risk management due diligence. This included designing risk mitigation into the device itself and including appropriate operating instructions and warnings into the device user manual. As medical devices transform into systems that include enterprise IT infrastructure provided by the hospital, risk management also becomes the responsibility of the customer.
Current industry best practice in clinical engineering limits risk management to determining electrical safety, optimal calibration, and preventive maintenance periods. In IT, formal risk management is focused on privacy and security. While risk management in both departments is focused on important topics, there is a conspicuous gap with regards to patient safety and the ongoing management and support of all the components that make up a medical device system.
A combined effort of clinical engineering and IT is needed to identify and mitigate risks associated with the configuration and ongoing enhancement of IT infrastructure that has become part of medical device systems. This needed risk management effort should guide formal and active configuration management and change control for both medical device systems and the IT infrastructure that supports them.
There are two standards that hospitals can use to guide risk management of medical device systems. In 2010, the IEC 800014 standard was promulgated to apply risk management for IT networks incorporating medical devices (Gee, 2008). Another more generalized standard for medical device risk management, and referenced by IEC 80001, is ISO 149715. Both of these standards provide a best practice foundation for medical device system risk management in hospitals.
Configuration Management
Configuration management is an activity that both clinical engineering and IT engage in daily. And while clinical engineering often has ad hoc solutions to configuration management for specific types of devices or systems (often received from manufacturers), there is seldom a formal policy and procedure that guides this process. Likewise in IT, configuration management of systems, applications, and infrastructure is often done on an informal basis. Some hospital IT departments use enterprise IT tools to automatically capture configuration data and to monitor the infrastructure for configuration changes. This is a best practice that should be extended to medical device systems and adopted by all hospitals.
Baseline system configurations for medical device systems should be captured at the time of initial installation after the medical device manufacturer has “certified” the installation as conforming to their specifications. Installation specifications can be acquired from medical device manufacturers for already installed systems to confirm they are within specification.
Because clinical engineering and IT converge in managing medical device systems, configuration management should also be a converged common process, with a common set of tools. How configuration management tasks are split between clinical engineering and IT is much less important than having a common process and tools that both groups use to document and manage medical device system configurations. Configuration management should be a proactive rather reactionary; it’s far better to anticipate problems rather than to react to problems that arise from a lack of configuration management. Best practice configuration management is driven and informed by a risk management process.
Change Control
Change control goes hand-in-hand with configuration management. As medical device and IT manufacturers update product software or firmware, release new products, and discontinue old ones, it is necessary to change portions of medical device systems. The challenge for medical device manufacturers is to accommodate those changes in a timely fashion while ensuring the safety and effectiveness of their medical device system. To accomplish this, manufacturers must follow their quality system to develop requirements, create a new design based on the required changes, and complete verification and validation testing to ensure the changes are safe and effective. At least two risk analyses are done during this process. The FDA requires the manufacturer to document the foregoing processes in detail. While not a common occurrence, depending on the scope of the update, the manufacturer may also be required to receive FDA clearance before the product update is released. The scope of effort can vary based on the type and degree of change required; most product updates or fixes can be engineered, tested, and released in a timely fashion.
Common causes for medical device changes or fixes include eliminating a latent software defect, correcting a feature to ensure proper operation, or patching a component’s operating system (common for Windows-based medical devices). A more frequent source of changes comes from the off-the-shelf (OTS) system components used in medical device systems such as WiFi radios and access points, network switches and routers, servers, and virtualized environments. When these OTS components are updated or discontinued by their original manufacturers, medical device manufacturers must assess the impact, starting with a risk analysis. Any design changes required to support the revised or new model of OTS component must be completed, tested, and released. This is the point at which the hospital receives a product update and must plan their change control process.
Once changes are released by the medical device manufacturer, it is the hospital’s responsibility to evaluate the implementation in its environment. This evaluation and resulting execution is the heart of change control in the hospital. Change control as implemented by clinical engineering is often limited to simply applying upgrades and patches supplied by the medical device manufacturer. In IT, change control is a more formal process that looks beyond the upgrade or patch at hand and considers the impact of the change on the broader infrastructure. Rarely is the broader impact extended to formally include medical device systems.
A potential complexity for hospitals can arise when changes to medical device systems, or other IT systems, result in configurations that conflict with other medical device or IT systems. Resolving these conflicts can be complex and time consuming. Successful change control of medical device systems requires clinical engineering and IT to utilize a harmonized set of policies and procedures (or the same policies and procedures) to ensure sufficient rigor to maintain safe and effective medical device systems.
Adequate Test Environments
The complexity of systems and systems-of-systems6, combined with the patient safety risks inherent with medical device systems, requires a robust environment in which to test proposed changes before they “go live” for use with patients. To be effective, test environments must physically duplicate or simulate the actual production environment in which systems operate. Test plans are then developed to exercise that environment to ensure the system operates as expected. Best practice test environments duplicate production environments where practical and simulate production environments where a full duplication is too resource intensive. A risk analysis is the best method for determining if a proper test environment is sufficient for the task of testing a particular system, upgrade, or patch.
The typical hospital IT test environment is a mixed bag, meeting requirements in some areas and generally falling short in others. Most IT test environments for servers are very adequate for testing safety-critical systems. Server test environments do not require much physical space or hardware, so it is simple to create an environment that duplicates or simulates actual production server environments.
Duplicating networks or broader information or medical device systems is much more challenging. Here it is often impractical to create a test environment that duplicates the target environment. Duplicating entire systems is also difficult. As with networks, simulation is critical to creating a test environment that provides a technically meaningful representation of the system to be tested.
The degree of network and system testing (beyond sever testing) in most hospitals is minimal. Often network changes and system updates are “tested” in phased deployments in the actual production environment. The limitation of this approach can be seen in frequent problems resulting from network changes in hospitals. This approach falls considerably short of a testing regimen that meets safety-critical system requirements.
Summary
Besides the continued automation of diagnostics, monitoring, and therapy delivery at the point of care, additional applications such as remote ICUs and remote anesthesiology are transforming healthcare IT into a safety-critical endeavor. The days of healthcare IT crunching data at a level of risk and reliability equivalent to industries such as finance, logistics, or even manufacturing are fading. Clinical engineers have always worked in an environment where mistakes can result in patient injury or death. Safety-critical applications are a new thing for IT, something most are just coming to realize.
The current level of collaboration between clinical engineering and IT must be improved upon, to include a holistic and rigorous framework of policies and procedures to realize true safety-critical operating levels for both medical devices and IT. Many of the right things are being done on an informal level. Yet to ensure consistent and safe results, both clinical engineering and IT must adopt a new safety-critical systems approach. Job titles and reporting structures are secondary to what is done and how it is done. I hope this article provides a starting point for assessing your organization’s readiness for safety-critical systems support and a path for adopting evolving best practices. ‚ùô
Tim Gee is principal and founder of Medical Connectivity Consulting (www.medicalconnectivity.com) where his practice revolves around workflow automation through the integration of medical devices with information systems and enabling technologies. Gee is the 2012 recipient of the American College of Clinical Engineering’s Challenge Award and is program chair and principal organizer for the Medical Device Connectivity conference. Gee writes at the Medical Connectivity blog, serves on the editorial advisory boards of a number of publications including Patient Safety & Quality Healthcare, and participates in key industry interoperability development efforts. He may be contacted at tim@medicalconnectivity.com.
|
The shortcomings described in this article do not result in sentinel events on a frequent basis. Neither do they represent best practice for safety-critical systems. Besides the foregoing information in this article, hospitals can use the following series of questions to quickly assess any gaps in managing medical device systems: 1. Is there more than one inventory management system used to document and manage medical device systems and medical device software, including IT components? 2. Does clinical engineering have documented policies and procedures for documenting and managing system and module configurations? 3. Does clinical engineering apply risk management beyond determining electrical safety and recalibration/preventive maintenance periods? 4. Does IT apply risk management beyond privacy and security concerns? 5. Does IT have documented policies and procedures for documenting and managing configurations? 6. Does clinical engineering have a documented change control policy and procedure for medical device systems? 7. Does IT have a documented change control policy and procedure for medical device systems? 8. Is there a test lab where testing is done on medical device systems that include medical devices (or simulated medical devices) and associated IT infrastructure? Any “no” response to the above questions indicates a gap that should be filled. 7 To meet this requirement the test lab must have more than one controller, a couple of access points, and a router or switch. An adequate test environment will have the ability to test mobility (roaming across access points and subnets) and simulate data traffic in volume and from devices/systems that are representative of the hospital’s enterprise network environment. |